Peptide Quality Control: How Mass Spectrometry Confirms Research Compound Identity
For researchers working with peptides, confidence in compound identity is foundational to valid experimental results. While HPLC purity analysis confirms that a peptide preparation is free from significant impurities, it cannot definitively confirm that the major peak is actually the intended compound. A peptide could be highly pure by HPLC yet be the wrong sequence, contain an unexpected modification, or have an incorrect disulfide bond arrangement. This is where mass spectrometry (MS) becomes indispensable.
This guide explains how mass spectrometry is used to confirm peptide identity, how to interpret mass spectral data, and what researchers should understand about the capabilities and limitations of this essential quality control technique.
Why Molecular Weight Confirmation Matters Beyond HPLC Purity
HPLC separates compounds based on their interactions with a stationary phase, typically reversed-phase C18 media for peptides. The resulting chromatogram shows peaks corresponding to different species in the sample, and purity is calculated from peak area ratios. However, HPLC has inherent limitations for identity confirmation:
- Co-elution: Different peptides with similar hydrophobicity profiles can elute at the same retention time
- Isomeric species: Peptides with D-amino acid substitutions, deamidation products, or sequence inversions may be chromatographically indistinguishable from the target compound
- No structural information: HPLC retention time alone does not reveal molecular weight, sequence, or modification status
Mass spectrometry addresses these limitations by providing the molecular weight of the compound, which serves as a molecular fingerprint. Each unique peptide sequence produces a characteristic mass that can be compared against the theoretical value calculated from the sequence. The International Council for Harmonisation (ICH) guideline Q6B specifically recommends mass spectrometric characterization for peptide and protein identity testing.
ESI-MS: Electrospray Ionization Mass Spectrometry
Electrospray ionization (ESI) is the most commonly used ionization method for peptide analysis and is the technique most researchers will encounter on certificates of analysis from suppliers like Aureum Peptides.
How ESI Works
The ESI process converts peptides from solution into gas-phase ions through several steps:
- The peptide solution is passed through a narrow capillary held at high voltage (typically 2-5 kV)
- The electric field generates a fine spray of charged droplets at the capillary tip (a Taylor cone)
- As the solvent evaporates, the droplets shrink and the charge density increases
- Eventually, individual peptide ions are released into the gas phase and enter the mass analyzer
Multiply Charged Ions
A distinctive feature of ESI is that it produces multiply charged ions. A peptide can pick up multiple protons during the ionization process, resulting in ions with charges of +2, +3, +4, or higher. This is important for spectral interpretation because the mass spectrometer measures mass-to-charge ratio (m/z), not mass directly.
For a peptide with molecular weight M:
- [M+H]+ (singly charged): m/z = M + 1.008
- [M+2H]2+ (doubly charged): m/z = (M + 2.016) / 2
- [M+3H]3+ (triply charged): m/z = (M + 3.024) / 3
The presence of a characteristic charge state envelope (a series of peaks at predictable m/z values) provides additional confirmation of the molecular weight, since all charge states must yield the same calculated M value.
MALDI-TOF for Larger Peptides
Matrix-Assisted Laser Desorption/Ionization Time-of-Flight (MALDI-TOF) mass spectrometry is particularly well-suited for peptides in the higher molecular weight range and offers complementary capabilities to ESI.
Principles of MALDI
In MALDI analysis:
- The peptide sample is co-crystallized with a UV-absorbing matrix compound (commonly alpha-cyano-4-hydroxycinnamic acid for peptides)
- A pulsed laser irradiates the crystal, causing rapid matrix ablation that carries peptide molecules into the gas phase
- MALDI predominantly produces singly charged ions [M+H]+, simplifying spectral interpretation compared to ESI
- The time-of-flight analyzer separates ions based on the time required to traverse a field-free drift tube, with lighter ions arriving at the detector first
When MALDI Is Preferred
MALDI-TOF offers specific advantages for certain peptide analysis scenarios:
- Higher mass peptides: MALDI handles peptides above 5,000 Da more readily than some ESI configurations
- Simpler spectra: The predominance of singly charged ions makes interpretation straightforward
- Salt tolerance: MALDI is generally more tolerant of buffer salts than ESI, requiring less sample preparation
- High throughput: Multiple samples can be spotted on a single MALDI plate for rapid sequential analysis
How to Read a Mass Spectrum
Understanding a peptide mass spectrum requires comparing observed data against theoretical expectations. Here is a systematic approach.
Step 1: Calculate the Expected Molecular Weight
Using the peptide’s amino acid sequence, calculate the monoisotopic mass (using the most abundant isotope of each element) or the average mass (using isotope-weighted average atomic masses). Most peptide mass calculators provide both values. For peptides under approximately 4,000 Da, the monoisotopic mass is typically used; for larger peptides, the average mass is more practical.
Account for any modifications:
- Acetylation: adds 42.01 Da
- Amidation: subtracts 0.98 Da (replaces -OH with -NH2)
- Disulfide bonds: subtract 2.02 Da per bond (loss of two hydrogens)
Step 2: Identify the Major Ion
Locate the most abundant peak in the spectrum and determine whether it corresponds to the expected m/z for any charge state. For ESI spectra, check for the [M+2H]2+ ion first, as this is often the base peak for peptides in the 1,000-3,000 Da range.
Step 3: Confirm with Additional Charge States
If the spectrum is from ESI, verify that multiple charge states are present and that all yield the same deconvoluted molecular weight. Consistent molecular weight from independent charge states provides strong identity confirmation.
Step 4: Evaluate Mass Accuracy
The observed mass should match the theoretical mass within the instrument’s specified accuracy. Typical expectations:
- High-resolution instruments (Orbitrap, Q-TOF): Within 5 parts per million (ppm)
- Standard ESI instruments: Within 0.1-0.5 Da
- MALDI-TOF (linear mode): Within 0.1-0.5% of the molecular weight
- MALDI-TOF (reflector mode): Within 20-50 ppm
Common Mass Spectrum Artifacts
Researchers reviewing mass spectral data should be aware of several common features that can complicate interpretation.
Sodium and Potassium Adducts
Peptides readily form adducts with alkali metal cations present in solvents and labware. These appear as peaks at:
- [M+Na]+: M + 22.99 Da (sodium adduct)
- [M+K]+: M + 38.97 Da (potassium adduct)
These adduct peaks are not impurities; they represent the same peptide associated with different cations. Their presence is normal and does not indicate quality problems.
Matrix Clusters (MALDI)
In MALDI spectra, peaks from the matrix compound and its clusters appear in the low mass region (typically below 500-700 Da). These can be ignored when analyzing peptide peaks at higher m/z values.
In-Source Fragmentation
Excessively energetic ionization conditions can cause peptide fragmentation within the ion source, producing peaks at lower m/z values than expected. While this can complicate identity confirmation, it can also provide partial sequence information.
What Mass Spectrometry Can and Cannot Tell You
Researchers should understand the boundaries of mass spectrometric identity confirmation.
MS Can Confirm
- Molecular weight matches the expected value for the target peptide
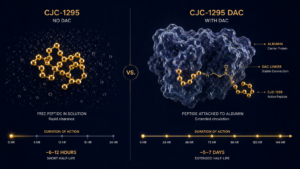
- Presence of expected modifications (acetylation, amidation, PEGylation, DAC conjugation for compounds like CJC-1295 DAC)
- Absence of major truncation products or gross synthetic errors
- Disulfide bond formation (by mass shift)
MS Alone Cannot Confirm
- Complete amino acid sequence: Simple MS measures mass only; tandem MS (MS/MS) is needed for sequence verification
- Stereochemistry: D- and L-amino acid substitutions produce identical masses
- Disulfide connectivity: Which cysteines are paired requires additional experiments
- Three-dimensional structure: Mass spectrometry does not assess folding or conformation
For comprehensive quality assurance, the ICH Q6B guidelines recommend combining mass spectrometry with complementary techniques including HPLC, amino acid analysis, and where appropriate, peptide mapping via tandem MS.
What to Look for on a Certificate of Analysis
When reviewing the certificate of analysis (COA) for research peptides from Aureum Peptides, the mass spectrometry section should include:
- Method used: ESI-MS or MALDI-TOF
- Observed molecular weight: The experimentally determined mass
- Expected molecular weight: The theoretical mass calculated from the target sequence
- Mass accuracy: The difference between observed and expected values, confirming they are within acceptable tolerance
A peptide that passes both HPLC purity criteria and mass spectrometric identity confirmation provides researchers with strong confidence that the compound is suitable for rigorous laboratory investigation.
Disclaimer: All products sold by Aureum Peptides are intended for laboratory and research use only. Not for human consumption. All peptides referenced in this article are sold as research chemicals. No statements on this page have been evaluated by the FDA. This product is not intended to diagnose, treat, supports research into, or may modulate any disease. For Research Use Only.